

Are you looking for research, analysis, or mandatory reporting services?
NAVIPPRO uses multiple data sources to assist with your research data needs.
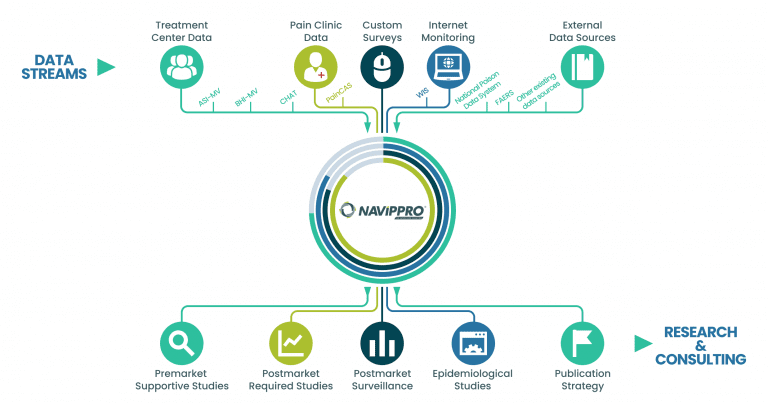

Treatment Center Data
We capture self-administered intake assessments from individuals being evaluated for substance use problems, allowing for rapid analysis of a large amount of data on a special population. These data can be used to demonstrate changes in substance use trends and may provide evidence for the potential abuse-deterrent characteristics of a prescription medication.
Using these data, we can also track trends of product-specific relative prevalence of nonmedical use, route of administration, and diversion of prescription opioid and stimulant products.

Pain Patient Data
We collect patient reported benefit and risk data from patients treated for pain. These data allow researchers to characterize patients treated for pain, their clinical presentations, treatments, medications, opioid misuse/abuse risk, and outcomes across geographic locations.

Custom Studies
Our experienced scientists partner with clients to design and conduct custom studies based upon the pressing clinical, regulatory, or commercial needs of your business. Each study is designed to address our client’s specific research questions and may cover a variety of topics, including product-specific questions about prevalence of nonmedical use, utilization patterns, diversion, and associated behaviors and outcomes.

Internet Monitoring
We capture and analyze data from a select number of publicly available recreational drug abuse discussion forums. With this historical and current data, Uprise Health captures trends over time regarding a variety of drug abuse-related topics and examines changes that occur when drugs are modified. These same validated methods can be applied to other consumer or prescription products of interest.

External Data Sources
We synergize complimentary data sources, integrated drug safety reports, or any other relevant data sources whether publicly or privately available to help meet clinical, regulatory, or commercial objectives.

Postmarket Required Studies and Surveillance
Our team of experts offers protocol development through final study report for postmarket required studies along with real-time postmarket surveillance for numerous prescription medications with abuse potential. We use a mosaic state-of-the-art approach tailored to our client’s regulatory requirements to ensure compliance.

Epidemiological Studies
We work closely with clients to tailor exploratory or hypothesis-driven epidemiological studies, including:
- Assessing the impact of substance use and chronic pain treatment protocols on patient populations locally, regionally, and nationally
- Understanding current trends to inform pre-market activities and supplement New Drug Applications
- Evaluating effectiveness of abuse deterrent formulations (ADFs) using the mosaic approach
- Tracking or comparing real-world prescription drug use trends from different populations of interest
- Understanding behavior associated with nonmedical use of prescription drugs, such as route of administration and source of drug acquisition

Publication Strategy
Our team of widely published scientists create strategic plans for dissemination of research findings. From conference presentations to peer-reviewed manuscripts to continuing medical education programs, we work to share key findings accurately and effectively with targeted audiences.